Language:
Japanese
English
PoTHoS
Phyto Transcriptomics by High-throughput Sequencer
Project
Top
Top page of project.
Settings
Configuration of project.
Register members
Control members who could access to data.
Sample
Register files
Submit fastq files (*.fastq.gz) for analysis.
Register samples
Group files into samples.
Register groups
Group samples.
系列登録
登録したサンプルまたはグループを系列にします。
Quality of sample
Display the number of reads derived from the in-line control.
Assemble
Settings
Settings for the assembling of reads.
Result
Summary of the assemble.
Detail of contig
Display the sequence of contig.
Search contigs
Search contigs by using BLAST.
Detect SNPs
Display SNPs observed in contigs.
Annotation
Settings
Configure annotation options.
Result
Summary of Annotation.
Mapping
Settings
Settings for the mapping the reads to the assembled reference.
Summary of Mapping
Show the number of reads, rate for mapping by each sample.
Expression level
Show tag count for each contigs.
Display the mapped image (pile image)
Draw pile image for the reads mapped to contig.
Two samples analysis
比較解析
サンプル間で発現量の比を計算し、遺伝子を絞り込みます。
Scatter plot
Draw scatter plot.
Multi samples analysis
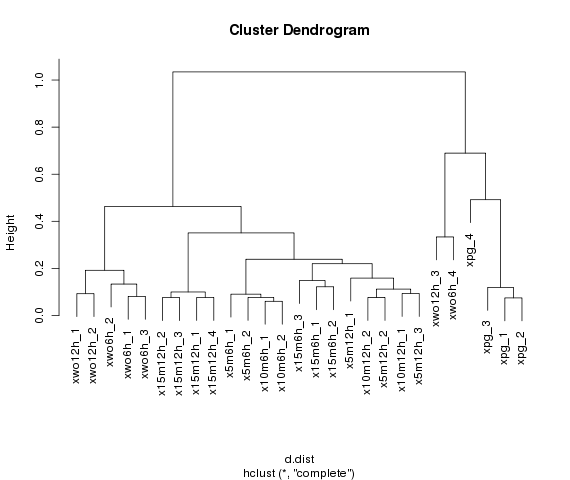
Cluster analysis
Make a cluster of samples based on the expression pattern of contigs.
Two groups analysis
edgeR
Differentical analysis between two groups using edgeR, an R package.
Reference
Nucleotide sequences
Search transcript sequence.
Protein
Search amino acid sequence.
Genome
Show genome sequence.
Blast search
Search sequences registered in PoTHoS by BLAST.
キーワード検索
配列IDまたはアノテーション中の文字列を検索します。
Download
Download nucleotide and amino acid sequences stored in PoTHoS as FASTA format.
Authentication
Login
Please login to show the data under access control.
Registration
Register e-mail address.
Cluster analysis among samples
Configure samples for cluster analysis
Please drag and drop samples for cluster analysis to used box.
Use all samples
Reset